The genome, made up of DNA, contains not only protein-coding sequences but also a vast array of non-coding regulatory sequences. These two types of sequence work in concert to shape the complex phenotypic traits of organisms, telling the ancient genetic stories of life. Deciphering the regulatory code hidden in this vast genetic landscape has long been a scientific summit that researchers strive to reach.
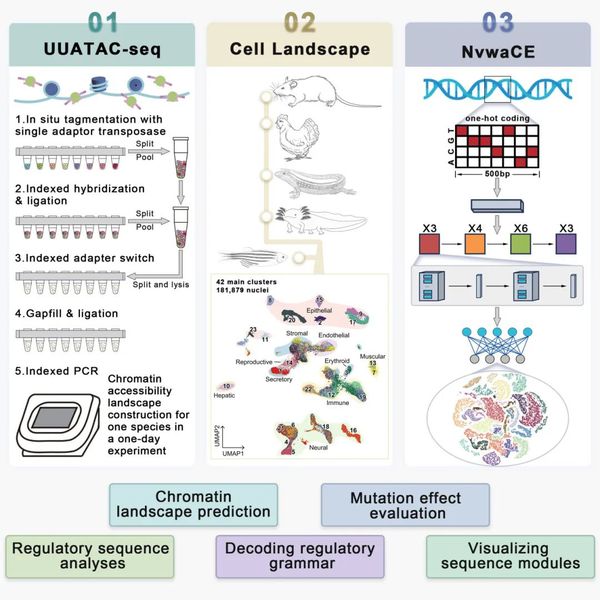
A team led by Professor GUO Guoji at Zhejiang University has taken a major step toward that goal. Building on their UUATAC-seq technology, an ultra-high-throughput, ultra-sensitive single-nucleus ATAC sequencing method, the team has constructed a vertebrate cCRE landscape and developed a multi-task deep learning model named NvwaCE. This model can directly predict single-cell regulatory sequence landscapes from genomic sequences. Their study, titled “Modeling the vertebrate regulatory sequence landscape by UUATAC-seq and deep learning”, was published in Cell on July 8.

Since the Human Genome Project began in 2003, which brought together the world's top scientists to map the human genome, less than 10% of the genetic information has been deciphered. How can researchers accelerate our understanding of such a complex system? In recent years, artificial intelligence (AI) has emerged as a powerful new ally.
For AI models to perform well, the quality of their training data — their “textbooks” — is of vital importance.
“The real challenge for genomic AI models, unlike structural AI models, lies in the pronounced batch effects of published data,” explains GUO. His team, with years of expertise in single-cell omics, previously used their Microwell-seq high-throughput single-cell mRNA sequencing platform to publish the world's first mouse and human cell atlases. Building on this experience, they developed UUATAC-seq as a next-generation ultra-sensitive method.

Unlike traditional approaches, UUATAC-seq captures the regulatory DNA that determines mRNA expression. This method provides epigenetic regulatory insights that complement transcriptomic data, while also avoiding missing low-expression genes such as transcription factors. This leads to improved data quality, more comprehensive information, and the capacity to profile a species' chromatin accessibility within a single day.
“Regulatory sequences are like functional switches in the genome. They need to be in an open, relaxed state to work,” GUO explains. Because different cell types have different regions of open chromatin, mapping chromatin accessibility is akin to drawing a functional blueprint of the genome, laying the foundation for deep learning models to understand the genetic “language” in coding different cell types.
Using UUATAC-seq, the team generated whole-body single-cell chromatin accessibility maps for five representative vertebrates: mouse, chicken, gecko, axolotl, and zebrafish. From this data, they identified millions of candidate cis-regulatory elements (cCREs) and systematically revealed the cell-type-specific regulatory programs that span vertebrate evolution.
“We found that the regulatory syntax of vertebrates is far more conserved than the nucleotide sequences themselves,” GUO notes. “This syntax classifies regulatory elements into different functional modules in high-dimensional space,” offering new insights into the sequence basis of cell-type-specific gene expression.
With this high-quality “textbook,” the team trained their deep learning model NvwaCE, which has become a powerful tool for decoding the regulatory logic of genomes. NvwaCE has learned to predict chromatin accessibility at the single-cell level from raw DNA sequences—not just for species it was trained on, but also for entirely new vertebrates, thanks to its impressive generalization capabilities. Notably, NvwaCE's prediction of human regulatory element accessibly strongly correlates with experimental measurements, showcasing its reliability.
“NvwaCE outperforms existing genomic AI models on multiple metrics,” says GUO. “It can accurately predict the functional impact of synthetic mutations on lineage-specific regulatory sequences.” This model can not only forecast phenotypic changes caused by mutations in specific cell types, but also identify potential therapeutic targets based on disease phenotypes.
So how does NvwaCE perform in real-world scenarios?
In one functional experiment, it predicted HBG1-68:A>G as a therapeutic target for sickle cell anemia. “Gene editing at this site significantly boosted fetal hemoglobin expression, compensating for the loss of function in β-hemoglobin caused by this sickle cell disease,” GUO explains. This marks the world's first experimentally validated, AI-designed therapeutic site for a human disease, a milestone that lays the groundwork for the future decoding of the genomic language and the construction of digital life models.
Compared to international counterparts, NvwaCE is trained on the highest-quality single-cell atlas data to date, achieving an AUROC > 0.90 for nearly all cell types, a level of predictive accuracy unmatched by most current genomic AI model.

“This study not only provides a valuable cross-species single-cell data resource, but also delivers a powerful AI tool for genome prediction,” GUO concludes. With its ability to decode regulatory logic, uncover the mechanisms of genetic diseases, and assist in designing synthetic regulatory sequences, NvwaCE is poised to become a vital resource in life sciences, medicine, and agriculture.
Adapted and translated from the article by ZHA Meng
Translator: FANG Fumin
Photos by ZHE Yin
Editor: HE Jiawen, ZHU Ziyu

Bird flu, SARS, HIV/AIDS … Many of the infectious diseases that have unsettled the world have one thing in common: they first emerged in animals. Birds, bats, monkeys and other hosts can carry viruse...

A large fraction of the human genome is built from the remnants of ancient viral infections. Long dismissed as genetic clutter, these sequences are increasingly being implicated in early development. ...
G protein–coupled receptors (GPCRs) are among the most important “signal receivers” in the human body, translating the external stimuli into internal signals that govern sensation, mood, cardiovascu...