Hypertrophic cardiomyopathy (HCM), characterized by left ventricular hypertrophy and fibrosis, is the most common monogenic cardiovascular disorder with global distribution and diverse clinical spectrum. The disease prevalence has been estimated at approximately 1:500 in the general population and even higher (1 case per 200) when both clinical and genetic diagnoses are taken into accounts. Patients with HCM are predisposed to heart failure, outflow tract obstruction and arrhythmias. The most visible and feared consequence has been the increasing risk of sudden cardiac death (SCD). HCM has been repeatedly regarded as the most common cause of SCD in young people and athletes. MYBPC3 is the most frequently mutated gene in HCM, accounting for more than 50% of cases in which the causative gene has been identified. Full-length MYBPC3 haploinsufficiency has been considered as the possible mechanism underlying the pathogenesis of HCM. But the exact mechanism by which MYBPC3 mutations lead to HCM is not resolved.
The findings by Professor LIANG Ping’s Lab at Zhejiang University School of Medicine, which were published online December 31 in Clinical Translation Medicine, present accelerated HSC70-mediated MYBPC3 protein degradation as a novel mechanism of enhanced diastolic Ca2+ leak from ryanodine receptor 2 (RYR2) in HCM, leading to hypertrophy and aberrant Ca2+ handling. “This study will be helpful for elucidating the molecular mechanisms underlying MYBPC3-related HCM and for identifying novel therapeutic drugs for the disease,” said Prof. Liang.
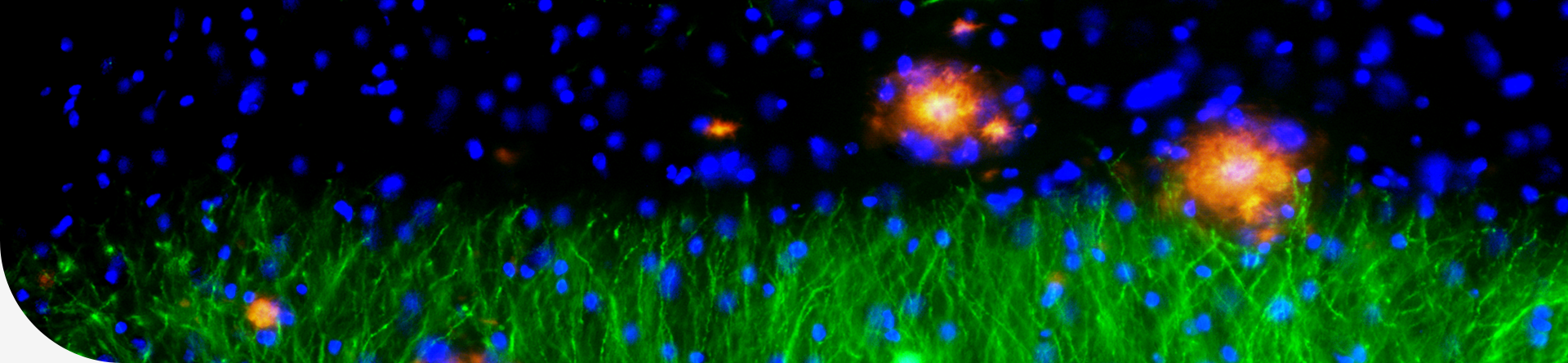
In this study, the researchers identified a MYBPC3 mutation in four unrelated individuals with HCM. To assess the pathogenicity of the variant and pinpoint underlying mechanisms, wild type (WT) or mutant MYBPC3 was introduced in healthy induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs). With angiotensin II (Ang II) treatment, mutant iPSC-CMs exhibited a variety of deleterious phenotypes, including reduced MYBPC3 expression, hypertrophy, arrhythmia and elevated diastolic intracellular Ca2+ [Ca2+]i. Moreover, RNA sequencing of mutant iPSC-CMs revealed a common gene characteristic of HCM and upregulated HSC70 expression. In iPSC-CMs, HSC70 accelerated protein degradation of MYBPC3 via lysosomal pathway under Ang II stress, and inhibition of HSC70 restored the MYBPC3 expression and alleviated hypertrophy and Ca2+ handling abnormalities caused by the mutation. The reduced MYBPC3-binding ryanodine receptor 2 (RYR2) caused by insufficiency of MYBPC3 protein may give rise to excessive free destabilized RYR2, which in turn caused larger RYR2-mediated Ca2+ leak. The resultant elevated Ca2+ loading may trigger the development of both hypertrophy and arrhythmogenesis, particularly under stress conditions.
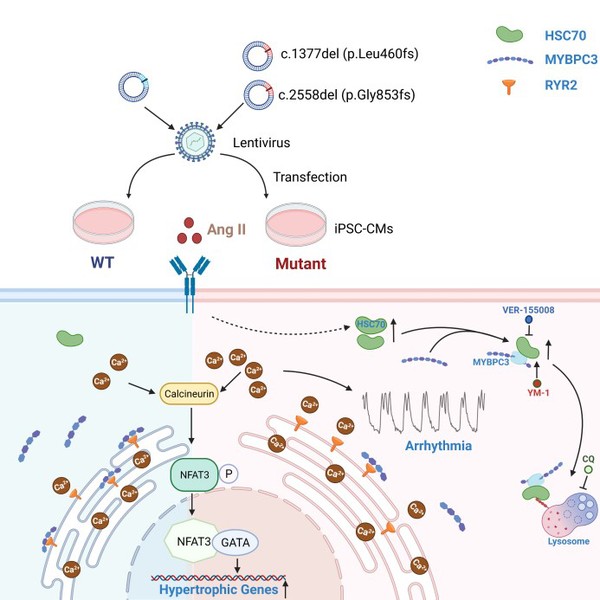
Proposed work model
This work has several novelties and clinical implications. Firstly, the study demonstrated that, under Ang II stress, iPSC-CMs carrying MYBPC3 mutation can recapitulate pathological features of HCM in vitro. We provide robust evidence to support that the MYBPC3 variant is a pathogenic mutation for HCM. Secondly, researchers showed that HSC70 plays a pivotal role in HCM development by regulating MYBPC3 protein turnover. The elevated HSC70 expression accelerates the MYBPC3 degradation via lysosomal pathway under Ang II stress, which may aggravate the haploinsufficiency of heterozygous MYBPC3 premature termination codon (PTC) mutation. Thirdly, the results suggested that MYBPC3 is not only a sarcomere protein for contraction force modulation, but also participates in regulating Ca2+ signaling. The deficiency of MYBPC3 protein intensifies the RYR2-mediated Ca2+ leak and causes elevated diastolic [Ca2+]i, which is essential for triggering hypertrophy and arrhythmia. Lastly, these findings will help elucidate the molecular mechanisms underlying MYBPC3-related HCM and identify a potential therapeutic target for the disease.
Source:LIANG Ping's Lab
Although cellular immunotherapy represented by CAR-T cells has achieved revolutionary success in the treatment of hematological malignancies, its therapeutic efficacy remains constrained by intrinsic ...

Bird flu, SARS, HIV/AIDS … Many of the infectious diseases that have unsettled the world have one thing in common: they first emerged in animals. Birds, bats, monkeys and other hosts can carry viruse...

A large fraction of the human genome is built from the remnants of ancient viral infections. Long dismissed as genetic clutter, these sequences are increasingly being implicated in early development. ...